
Нарушение эмбриогенеза на ранних стадиях приводит к тяжелым аномалиям развития плода. Немногие знают, что такое , синдром Ди Джорджи (или Ди Георга, велокардиофациальный синдром).
Это тяжелая генетическая патология, связанная с соматическими хромосомами.

Основные проявления – нарушение формирования вилочковой, паращитовидных желез, множественные пороки развития.
Причины появления синдрома
Биологической основой синдрома Ди Джорджи является делеция (или выпадение) участка длинного плеча 22 пары хромосом. В 9 из 10 случаев это происходит спонтанно в результате мутации.
У 1 из 10 патологическая хромосома наследуется от одного из родителей по аутосомно-доминантному типу. В исключительных случаях велокардиофациальный синдром проявляется вследствие перестройки в других хромосомах.
Этиопатогенез реализуется в выпадении участка хромосомы. Это приводит к потере важного белка, отвечающего за правильное формирование жаберных карманов зародыша. Генетическая патология вызывает:
| грубые аномалии сердца и отходящих от него сосудов, |
| незаращение твердого неба и верхней губы, |
| нарушение формирования тимуса и паращитовидных желез, |
| задержку нервно-психического развития. |
Частота обнаружения синдрома составляет до 1 на 3000 детей любого пола, родившихся живыми.
Клинические признаки
Путем генетических исследований было установлено, что у большинства пациентов с признаками синдрома Ди Джорджи выпадает один и тот же участок из 22 пары хромосом, однако разнообразие внешних проявлений патологии может варьировать даже среди ближайших родственников.
Выраженность симптомов, определяющих картину болезни, меняется с возрастом.
Часто велокардиофациальный синдром диагностируют вскоре после рождения. У грудных детей тяжесть состояния определяется наличием пороков сердца и нарушениями в составе крови вследствие гипопаратиреоза.
Затем на передний план выступает слабая сопротивляемость к инфекционным заболеваниям, аутоиммунные процессы и отставание в развитии.
Внешний вид пациента
Характерные фенотипические особенности не остаются незамеченными уже в первые месяцы после рождения.
Со временем клиническая картина становится более отчетливой:
|
|
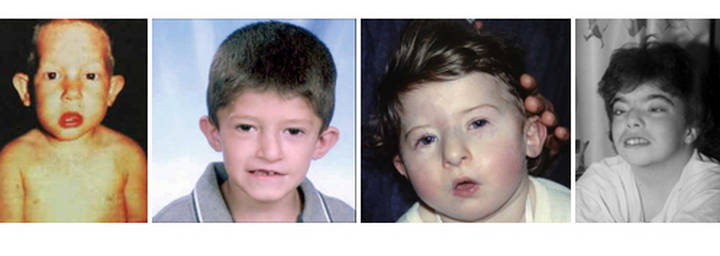
Патологии носоглотки
Нарушение развития глоточного кармана регистрируется более, чем у 60% пациентов и приводит к характерным стигмам и аномалиям носоглотки:
- расщелины нёба,
- высокое нёбо,
- расщелины верхней губы,
- нарушение обоняния,
- нарушение глотания,
- тугоухость.
Поражение сердца и сосудов
У 80% пациентов синдром Ди Георга характеризуется наличием пороков сердца и крупных сосудов. Чаще встречаются: тетрада Фалло:
- Сужение выхода из правого желудочка,
- Развитие гипертрофии правого желудочка,
- Отверстие в межжелудочковой перегородке,
- Правостороннее расположение дуги аорты.
Такая аномалия развития может сочетаться с прерыванием дуги аорты, наличием общего легочного ствола и другими дефектами.
Пороки сердца часто диагностируются во время планового УЗИ плода. В большинстве случаев такие аномалии требуют многочисленных оперативных вмешательств в первые месяцы жизни ребенка.
Недоразвитие паращитовидных желез
Гипопаратиреоз, сопровождающий велокардиофациальный синдром, возникает ввиду того, что паращитовидные железы у таких людей отсутствуют или недоразвиты.
Это нарушает всасывание кальция в кишечнике и его выведение почками. В результате концентрация вещества в крови падает, компенсаторно растет содержание фосфатов.
Возникает судорожный синдром.
Аплазия также приводит к:
- изменениям кожи,
- нарушению роста костей,
- патологии хрусталика.
Отсутствие или недоразвитие паращитовидных желез устанавливают при ультразвуковом обследовании человека.
С возрастом пациенты с велокардиофациальным синдромом обращают на себя внимание отставанием в росте и задержке в психическом развитии.
Интеллектуальная сфера страдает у 80% детей, во взрослом состоянии у них часто возникают психические нарушения.
Проявления аплазии тимуса
Врожденная аплазия или гипоплазия тимуса (отсутствие или грубое недоразвитие) регистрируется более чем у 70 % пациентов.
Эта патология заслуживает пристального внимания, так как приводит к первичному иммунодефициту. Он проявляется в следующем:
- угнетение первичного ответа на инфекцию,
- нарушение передачи информации о чужеродных агентах B-лимфоцитам,
- нарушение иммунной памяти,
- аутоиммунные патологии.
Недостаточность иммунитета можно заметить по состоянию здоровья: такие дети подвержены частым вирусным заболеваниям (чаще , дыхательной системы), возникают вторичные бактериальные осложнения, грибковые инфекции.
Недостаточность Т-хелперов при гипоплазии вилочковой железы вызывает нарушение координации иммунного ответа. Такие пациенты чаще подвержены аутоиммунным нарушениям.
Диагностические критерии заболевания
Рождение ребенка с характерными стигмами эмбриогенеза или дальнейшие нарушения развития являются показанием для обследования.
Проводят полный осмотр с выявлением аномалий развития, нарушения роста, дыхательной и сердечной недостаточности.
При необходимости привлекают следующих специалистов:
| эндокринолог, |
|---|
| кардиолог, |
| иммунолог-аллерголог, |
| челюстно-лицевой хирург, |
| невролог, |
| психиатр. |
Лабораторные исследования включают определение парат-гормона и гормонов щитовидной железы, содержания кальция в крови, концентрацию иммуноглобулинов и число Т-лимфоцитов.
Если ребенку делали прививки, то проверяют, сохранились ли антитела к специфическим инфекциям.
Инструментальные исследования включают в себя УЗИ сердца и внутренних органов, ЭКГ, рентген грудной клетки.
При поражении носоглотки возможно эндоскопическое обследование. Указание на патологию органа слуха требует проведения аудиометрии.
По показаниям выполняют МРТ, ангиографию.
Похожие внешние проявления характерны и для других заболеваний. Верифицировать диагноз синдрома Ди Джоджи помогают молекулярно-генетическое методы. Рекомендуют проведение тестов:
- определение мутаций в гене TBX1,
- тест методом флуоресцентной гибридизации (FISH) с ДНК-зондом,
- выявление делеции части плеча 10 пары хромосом.
Лечение патологии
Так как велокардиофациальный синдром – результат генетической аномалии, специфической терапии не существует. Лечение патологии направлено на профилактику или устранение угрожающих состояний.
Наиболее сложным считают период от рождения до 5 лет. В это время манифестируют клиника сердечной патологии, судорожный синдром, нарушение глотания, иммунодефицит, задержка развития.
Лечение заключается в операциях на сердце и сосудах, пластике нёба, зондовом кормлении, введении кальция.
Задержка речевого развития требует вмешательства невролога, логопеда. Выраженный иммунодефицит – показание к заместительной терапии.
В школьном возрасте синдром проявляется в виде гиперактивности, нарушения усвоения материала, частых инфекций и лечится противовирусными, антибактериальными препаратами, иммунотерапией, корректорами поведения.
После 18 лет ухудшение состояния связаны с нарушениями иммунитета и психическими расстройствами, лечение которых затруднено ввиду резистентности состояния больного к препаратам.
Прогноз для пациента
Прогноз при синдроме Ди Георга определяется тем, насколько пострадали основные функции организма.
В случае грубой патологии пациенты гибнут в раннем возрасте. При незначительных проявлениях клиника может определяться слабым иммунитетом и расстройствами поведения.
В таком случае пациенты живут достаточно долго, их удается социализировать.
Профилактика синдрома
Предотвратить рождение ребенка с этой генетической аномалией можно, если известно, что у кого-то из родителей есть делеция плеча 22 хромосомы.
Вероятность заболевания потомства составляет 50%.
После консультации генетика беременной женщине проводят амниоцентез и выполняют FISH-диагностику.
Однако в большинстве случаев мутация носит спонтанный характер, повлиять на вероятность ее развития невозможно.
К неинвазивным методам диагностики относят Panorama тест. Его проводят с 9 недели беременности, материал , кровь женщины.
Что делать при синдроме Ди Джорджа
Для своевременной диагностики этой аномалии следует выполнять скрининговые УЗИ плода. Подозрение на мутацию служит основанием для проведения амниоцентеза.
При выявлении синдрома Ди Джорджи у плода на сроках до 12 недель возможно проведение аборта по желанию женщины. В ряде случаев такие беременности не сохраняются.
На более поздних сроках вопрос о прерывании решается при выявлении тяжелых патологий у ребенка.
Если женщина планирует донашивать беременность, она должна быть в полной мере ознакомлена с последствиями своего решения и четко представлять себе, с какими отклонениями в состоянии здоровья ребенка ей придется столкнуться.